Free-Wilson edge-cases
I’ve always been a fan of Free-Wilson Analysis. It’s just such a useful tool: it’s easy-to-use, it helps you understand SAR better and it generates all potentially interesting combinations of your compounds that you might have missed. What’s not to love? In this post I’m going to address a few edge cases that the current code I’m using (written by the fantastic Pat Walters) can use a hand with.
Pat has written up a wonderful post on Free-Wilson Analysis over on Practical Cheminformatics accompanied by this notebook, and I’ll be using that very code as a starting point. You can find my notebook here if you’d like to follow along.
First, the edge cases. The current code uses molzip to combine all the fragments back into a single molecule. That works well for the most part, but it struggles with molecules which have two R-groups on the same attachment point, or rings that attach to two attachment points simultaneously. Molzip essentially wants to find exactly two attachment points that share an index: that’s how it knows which atoms to connect. I’ve drawn up three test cases where that is not the case, in order to verify that our improvements actually work:
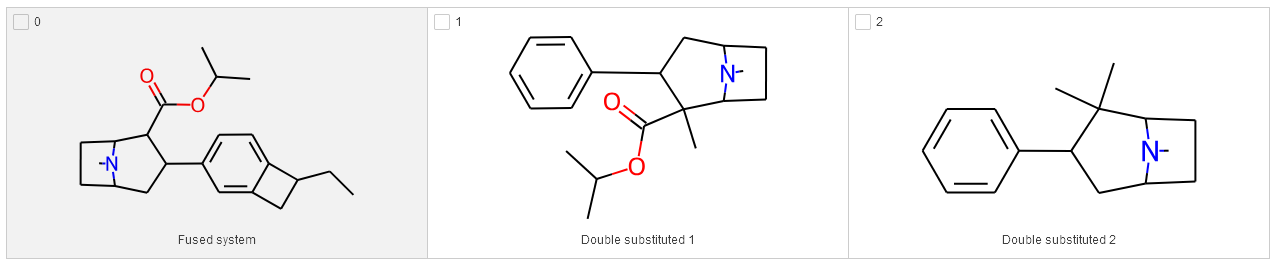
The first thing we need to fix is the way two R-groups on the same attachment point are handled. By default RDKit groups these into one attachment, separated by a period, e.g. C[*:3].C[*:3]. This causes trouble when we will be molzipping these compound back together later on, because we will be using the same attachment point twice which RDKit (rightfully) doesn’t appreciate. This snippet by Greg changes the default behaviour to create a second attachment point on the double-substituted attachment points. Rather than C[*:3].C[*:3], you should now get something like C[*:3] and C[*:4] in two separate R-group columns.
from rdkit.Chem import rdRGroupDecomposition
ps = rdRGroupDecomposition.RGroupDecompositionParameters()
ps.allowMultipleRGroupsOnUnlabelled = True
match, miss = RGroupDecompose(core_mol,df.mol.values,asSmiles=True, options=ps)
That fixes issues with the double-substituted compounds, but we still run into trouble if we have rings that attach to two attachment points. If we have a ring that attaches to R1 and R5, RDKit will put that moiety in both the R1 and R5. This is logical behaviour, but it doesn’t play nicely with molzip and the way we’re currently laying out the different R-groups. There’s two cases here that we will look at in more detail. Firstly, combining the ring CCC(C[*:1])[*:5] at R5 with another substituent at R5 (or on R1 with another R1, for that matter) doesn’t really work - there’s no sensible molecule we could make here, so the best we can do is to skip it with the following code snippet:
import re
def has_shared_number(string_tuple):
"""Checks if any number within the [*:number] format is shared among strings in a tuple.
Args:
string_tuple: A tuple containing strings with potential [*:number] patterns.
Returns:
True if any number is shared, False otherwise.
"""
all_numbers = []
for string in string_tuple:
numbers = [int(match.group(1)) for match in re.finditer(r"\[\*:(\d+)\]", string)]
all_numbers.extend(numbers)
return len(set(all_numbers)) < len(all_numbers)
#Use this in our enumeration code
prod_list = []
for i,p in tqdm(enumerate(product(*enc.categories)),total=total_possible_products):
core_smiles = rgroup_df.Core.values[0]
if has_shared_number(p):
continue
try:
smi = (".".join(p))
mol = Chem.MolFromSmiles(smi+"."+core_smiles)
prod = Chem.molzip(mol)
prod = Chem.RemoveAllHs(prod)
prod_smi = Chem.MolToSmiles(prod)
if prod_smi not in already_made_smiles:
desc = enc.transform([p])
prod_pred_ic50 = full_model.predict(desc)[0]
prod_list.append([prod_smi,prod_pred_ic50])
except:
print(p)
break
This works in the sense that it finishes, but doesn’t let us use the fused ring in our enumerations anymore. This brings us to the second case: having the same ring fragment appear twice in R1 and R5, which does correspond to a sensible molecule here: in fact, this is the exact way such a molecule would get decomposed by our procedure. In our current iteration of the code it gets removed, so we need to rewrite it a bit to de-duplicate in the smiles joining step to avoid this issue.
prod_list = []
for i,p in tqdm(enumerate(product(*enc.categories)),total=total_possible_products):
core_smiles = rgroup_df.Core.values[0]
if has_shared_number(p):
if len(set(p)) != len(p):
smi = (".".join(list(set(p))))
else:
continue
else:
smi = (".".join(p))
mol = Chem.MolFromSmiles(smi+"."+core_smiles)
prod = Chem.molzip(mol)
prod = Chem.RemoveAllHs(prod)
prod_smi = Chem.MolToSmiles(prod)
if prod_smi not in already_made_smiles:
desc = enc.transform([p])
prod_pred_ic50 = full_model.predict(desc)[0]
prod_list.append([prod_smi,prod_pred_ic50])
And there we have it! Looking at our final enumeration results, we can see that the enumeration now finishes without failures and we can find our fused systems back in the results:
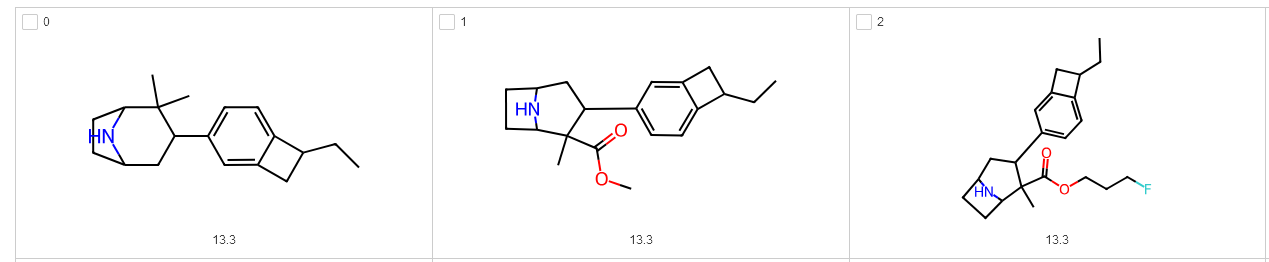
I hope this helps make your F-W explorations a bit more robust. Please let me know on Bluesky/X if you encounter any bugs!